by Ali Rind, Last updated: July 16, 2026

Sharing source documents with a pharma sponsor is not a routine file transfer. It is a regulated data disclosure. The patient records, lab results, consent forms, and visit reports inside those documents contain layers of protected health information (PHI), and removing that information correctly is a compliance requirement, not a formatting task.
Most clinical research teams reach for Adobe Acrobat or a similar general-purpose tool because it is already installed and familiar. These tools were not designed for the requirements of clinical trial document sharing. The gaps they leave are not edge cases. They are structural.
Why Redaction in Clinical Trials Is Different
When a CRO or NHS Trust prepares source documents for a sponsor audit or regulatory submission, the redaction task is specific. Patient identifiers must be removed completely, not visually obscured. Scanned records from paper-based systems must have handwritten and printed text detected and processed. The redaction decision must be traceable, with a record of who reviewed each page, what was removed, and when.
Compare this to general document handling, where most users need to hide a number or a name from a PDF before sending it internally. The tools built for that task are not built for this one.
The volume compounds the problem. A single Phase III trial can generate tens of thousands of pages. Manual review at that scale introduces inconsistency. One missed patient identifier in a sponsor submission is a GDPR breach. In a US-facing trial, it is a 21 CFR Part 11 violation affecting the integrity of the electronic record.
Where Adobe Falls Short
Adobe Acrobat has a redaction function. When used correctly on a text-based PDF, it does remove selected content permanently. The problem is that "used correctly" is a narrow condition that clinical trial workflows regularly fall outside of.
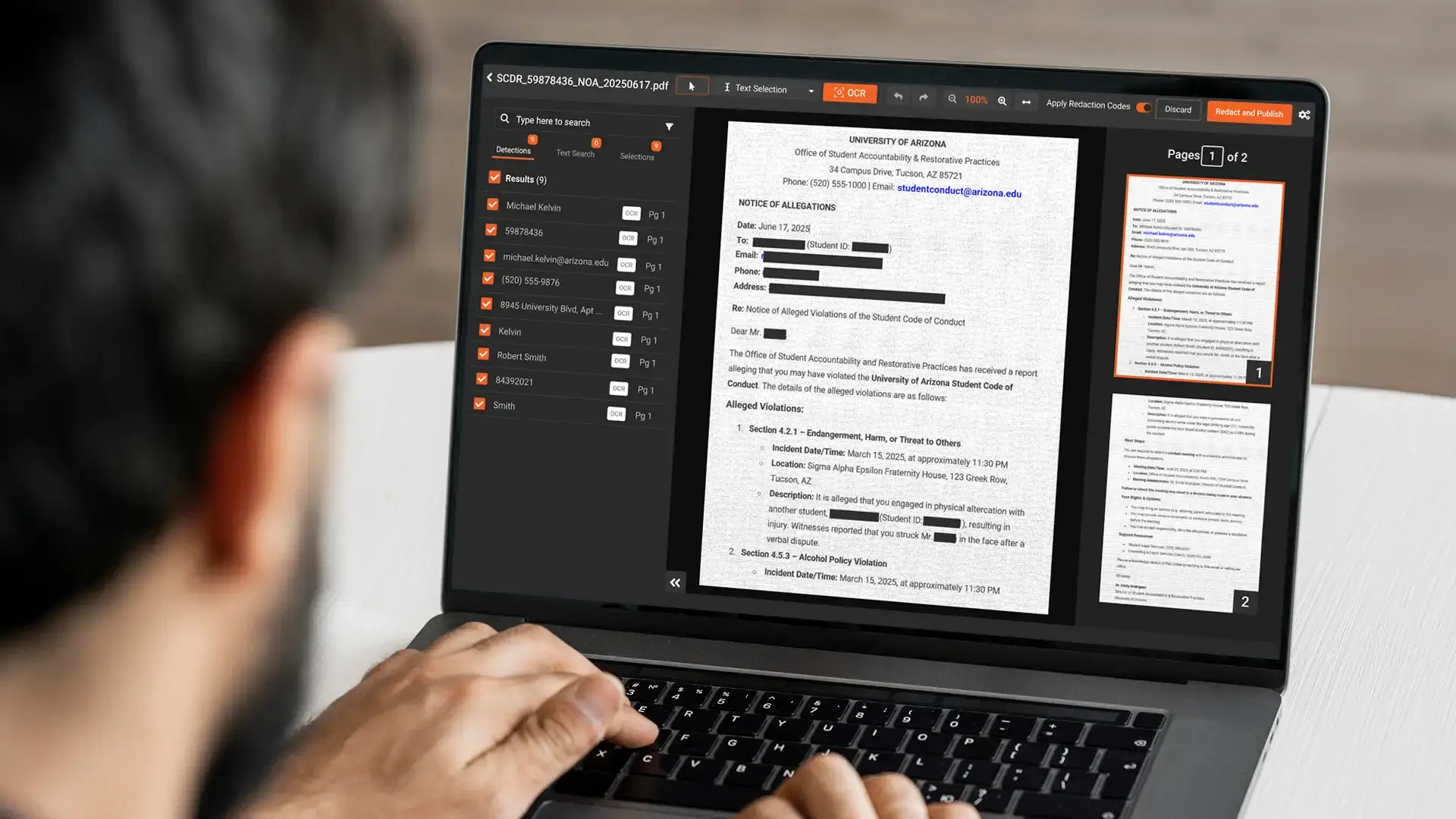
No PHI detection. Adobe does not scan for sensitive data. It executes whatever the user selects. In a document with 40 pages and a dozen patient identifiers scattered across headers, footers, tables, embedded images, and body text, a manual reviewer will miss some. There is no AI flagging what needs to go. The process depends entirely on individual attention, and individual attention fails at volume. For a broader look at what robust document redaction software should detect and handle, our overview covers the full capability set.
Scanned documents are not processed for PHI. Source documents from legacy hospital systems, paper consent forms, and handwritten lab notes arrive as image-based PDFs. Adobe can convert image to text via OCR, but it does not identify PHI patterns in that text. Someone still has to manually locate and redact every identifier across every page after conversion.
Metadata is not automatically removed. A redacted PDF still carries document properties, revision history, and author information. In a sponsor submission, that metadata is patient-adjacent data subject to the same compliance obligation as the content itself. Adobe does not strip it as part of the redaction workflow. This is a distinction covered well in our breakdown of data redaction vs. data masking and what permanent removal actually requires.
There is no audit trail. If a sponsor, data protection authority, or regulatory body asks how redaction decisions were made, who reviewed the document, and what criteria were applied, Adobe cannot answer that. There is no log of user actions, no record of what was detected and exempted, and no chain of custody.
For a one-off internal document, these gaps are manageable. For regulated clinical trial submissions, they are compliance failures.
What the Regulations Actually Require
ICH E6(R3) Good Clinical Practice guidelines establish that sponsors and investigators must protect trial participants' privacy and confidentiality, including in source documents shared with sponsor monitors and auditors. The standard does not specify a software tool, but it does require that processes are documented, controlled, and auditable.
Under GDPR and UK GDPR (which applies to NHS Trusts and UK-based CROs), processing participant personal data requires appropriate technical and organisational measures. A document with metadata intact, or where PHI was not detected because no detection was performed, does not meet that standard.
For US-facing trials, 21 CFR Part 11 governs electronic records and requires audit trails that capture who performed which actions and when. A redaction log is an electronic record under this standard.
The regulatory requirement is not just that PII is removed from the visible document. It is that the process of removing it was controlled, recorded, and defensible. Our guide on redaction software for legal and compliance workflows covers how these requirements translate into practical workflow controls.
What Purpose-Built Redaction Provides
VIDIZMO Redactor is built around the workflow gap that general-purpose tools leave open. The difference is in how redaction is structured: detection comes first, removal is permanent, and every step is logged.
The platform uses AI to detect PHI automatically across documents, including 40+ PII and PHI types: patient names, dates of birth, medical record numbers, health plan numbers, NHS numbers, and more. Detection runs before human review begins. Reviewers confirm and correct, rather than search from scratch. For clinical research teams handling medical records specifically, our walkthrough on how to redact PHI in medical records for clinical research shows the full workflow in practice.
For scanned source documents, OCR and handwritten text recognition run as part of the same pipeline. A paper consent form photographed and uploaded as a PDF goes through character recognition and PHI detection without a separate pre-processing step.
Redaction is permanent. Content is removed from the file structure. Metadata is stripped on output. The document that leaves the platform does not carry document history or author data from the original file.
Every action is logged. The audit trail records reviewer identity, timestamp, what was detected, what was redacted, and any exemption decisions. This log satisfies the documentation requirements of GDPR, UK GDPR, and ICH GCP, and provides a defensible record if a submission is questioned.
For organisations with high document volumes or regulatory-grade quality requirements, a managed redaction service with dual QA review is available. A second reviewer checks output before the document leaves the platform. This four-eyes approach is suited to the quality threshold of regulatory submissions.
The platform supports HIPAA with a Business Associate Agreement, making it appropriate for US-based trials and CROs operating across jurisdictions. For a full look at how the platform handles healthcare data redaction more broadly, including video and audio PHI, that resource covers the complete scope.
Talk to a redaction specialist about your clinical trial compliance requirements. See how VIDIZMO Redactor handles document redaction for sponsor submissions.

Key Takeaways
- General-purpose tools do not detect PHI. They rely entirely on manual selection, which means they rely entirely on manual accuracy.
- Metadata persistence in redacted PDFs is a compliance exposure that Adobe's standard workflow does not address.
- Scanned source documents require OCR-based PHI detection, not just character conversion.
- ICH GCP, GDPR, UK GDPR, and 21 CFR Part 11 all require documented, auditable redaction processes. A PDF editor does not produce an audit trail.
- Purpose-built redaction software handles detection, removal, metadata scrubbing, and audit logging as a single connected workflow, not as separate manual tasks.
People Also Ask
Adobe Acrobat's Redact tool permanently removes selected text when used correctly on a text-based PDF. The gap is detection: Adobe does not identify PHI automatically, does not process scanned documents for PII, and does not remove document metadata. In a clinical trial context, where volume is high and standards are regulatory, relying entirely on manual selection introduces systematic risk.
ICH E6(R3) Good Clinical Practice requires confidentiality protections and documented processes for source document handling. GDPR and UK GDPR require appropriate technical measures when processing participant personal data. 21 CFR Part 11 (for US-facing trials) requires audit trails for electronic records. None mandate a specific tool, but all require that the process is controlled and traceable.
Overlay redaction places a visual block over content without removing the underlying data from the file. The content remains in the file structure and can be extracted. Permanent redaction removes the data entirely. In clinical trial document sharing, permanent redaction with metadata removal is the appropriate approach to meet data protection obligations.
Yes. Purpose-built redaction platforms use AI to automatically detect patient identifiers, PHI, and other sensitive data types across documents, including scanned files. The AI flags candidate redactions for human review, reducing miss rate and processing time compared to fully manual approaches.
A defensible audit trail should capture reviewer identity, timestamp, what was detected, what was redacted, what was exempted and on what basis, and any QA review steps. The record should be tamper-proof and exportable for regulatory inspection.
About the Author

Jump to

Clinical Trial Redaction: A Practitioner's Guide for 2026
Best AI Redaction Software for Legal Teams in 2026

No Comments Yet
Let us know what you think